
Like many fifth graders, Natalie Van Wyk keeps busy after school with sports, clubs, piano lessons, and playdates with friends. But unlike her peers, Natalie has a social network that extends far beyond her north Texas hometown.
The 12-year-old has friends in Ireland, the United Kingdom, and Hungary who she can talk to about living with complications of a rare disease known as Generalized Arterial Calcification of Infancy (GACI). Caused by mutations in the ENPP1 or ABCC6 genes, GACI is a progressive condition that starts before birth with extensive buildup of calcium in the blood vessels—blockages that can ultimately cause heart attack, stroke, and death. At least 20 babies in the United States are born each year with GACI, a diagnosis that can be fatal for half within the first six months of life.
It’s a tragedy that Natalie’s parents, Jerry and Anne, know too well. Though they have three other living children—Drew, 20, and twins Julia and Graham, 15—Natalie’s older brothers, Reid Christopher and Ian James, were born with GACI and died within weeks. Determined that Natalie wouldn’t share the same fate, the Van Wyks started a journey in 2013 that would save their daughter’s life and lead them to Carlos Ferreira, M.D., a skeletal genomicist at the National Institutes of Health (NIH) and head of the Unit on Skeletal Genomics at the Eunice Kennedy Shriver National Institute of Child Health and Human Development (NICHD).
A trainee when he first met Natalie just months after her birth, Dr. Ferreira now leads research on rare genetic skeletal disorders from his lab in the NIH Clinical Center. By identifying the molecular mechanisms underpinning these conditions, he and his colleagues hope to understand skeletal growth and metabolism more broadly.
“There’s a lot that excites me about this field,” Dr. Ferreira said. “First, there are new technologies we can use to answer questions about the diagnosis or the mechanism of the rare disease we’re studying. Also, we can provide better medical care to patients because we can offer more targeted therapies.”

Of those born with GACI, up to 20% who survive infancy develop hypophosphatemic rickets by age two, and the vast majority develop the disease by adolescence. The condition causes bone pain, deformities, and short stature, among other complications. Additionally, those who have ENPP1 deficiency may experience early hearing loss as well as skin changes, retinal damage, and other musculoskeletal complications later in life.
When she came to NIH, Natalie was one of the first patients Dr. Ferreira and William Gahl, M.D., Ph.D., of NIH’s National Human Genome Research Institute, had seen with GACI. Building from previous studies and working with collaborators in Germany, they gradually amassed a cohort of more than 200 GACI patients in 19 countries, yielding the first natural history study of ENPP1 and ABCC6 deficiencies, which advanced the understanding of the disorder’s progression, prognosis, and symptoms.
For Natalie’s family, the NIH researchers’ focus on their daughter’s disease, progression, and treatments felt like a major step forward after years of struggle. “In our journey to have a family, we felt we were climbing uphill, and it was an insurmountable climb,” Jerry Van Wyk said. “So, for us to be invited to come to NIH to participate in any research, the answer was, ‘Absolutely, immediately, and what can we do?’”
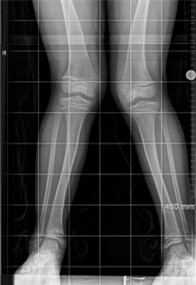
Credit: Genet Med. 2020 Oct 2;23(2):396–407
While scientists understand the molecular basis for why ENPP1 deficiency causes widespread arterial calcification and cardiovascular complications, they have not established why the deficiency later causes abnormal phosphate levels in GACI survivors, leading almost universally to rickets. Among other research, the Ferreira Lab conducts studies in cells and animals to understand the origins of rickets and the skeletal complications in patients with ENPP1 and ABCC6 deficiencies.
The Ferreira Lab monitors Natalie’s rickets, which started around age three and are managed closely with daily medications, such as phosphorus. They also collaborate with researchers at the Yale School of Medicine who developed an enzyme replacement therapy (ERT) that prevented arterial calcification and later complications in preclinical trials. Boston-based Inozyme Pharma is now using the therapy, known as INZ-701, in clinical trials for infants, children, and adults with rickets.
Though Natalie is not currently participating in ERT trials, GACI Global, a nonprofit organization founded by her parents and others, has been an essential resource for clinical trials and research. Founded in 2018, the group has connected families around the world who have children born with ENPP1 and ABCC6 deficiencies with information, clinical trials, and community support. “For years we thought we were all alone, so to see all that’s happened, and to have ERT trials happening, is mind-blowing to us,” Anne Van Wyk said.
As his team’s research on GACI and other skeletal diseases continues, Dr. Ferreira is hopeful that the growing body of knowledge about ENPP1 and ABCC6 deficiencies will yield uniform standards of care for better identifying and managing people with the condition. GACI continues to be underdiagnosed—only half of those born with the condition are accurately diagnosed—so early detection is critical. “One great way to improve the diagnostic accuracy would be newborn screening for GACI. Every baby born in the United States gets some screening.
Can we do a heel prick and analyze that blood? This is a dream right now,” Dr. Ferreira said.

Though she’s had a long medical journey, Natalie says her frequent visits to Dr. Ferreira and her contributions to research have had an unexpected upside: she’s considering being a nurse when she grows up. For her parents, working with Dr. Ferreira and doing what they can to advance rare disease research after heavy loss is invaluable.
“Participating in research at NIH has been an absolute privilege for us. Having NIH be on our side in fighting, combatting, and learning about this was like wind in our sails,” Mr. Van Wyk said.
 BACK TO TOP
BACK TO TOP