What is CAH?
CAH refers to a group of genetic disorders that affect the adrenal glands. These glands sit on top of the kidneys and release hormones the body needs to function. CAH is caused by three disturbances:
- Too little cortisol (pronounced KAWR-tuh-sawl). The adrenal glands of infants born with CAH cannot make enough of the hormone cortisol. This hormone affects energy levels, blood sugar levels, blood pressure, and the body's response to stress, illness, and injury.
- Too little aldosterone (pronounced al-DOS-tur-own). In about three-fourths of cases, infants born with CAH cannot make enough of the hormone aldosterone, which helps the body maintain the proper level of sodium (salt) and water and helps maintain blood pressure.1
- Too much androgens (pronounced AN-druh-juhnz or AN-druh-jenz). In certain cases, infants born with CAH produce too much of male hormones, androgens. Proper levels of these hormones are needed for normal growth and development in both boys and girls.
CAH can also cause imbalances in the hormone adrenaline (pronounced uh-DREN-uhl-in), which affects blood sugar levels, blood pressure, and the body's response to stress.2,3
The hormone imbalance most often seen in CAH cases is too little of a substance called 21-hydroxylase (pronounced hahy-DROK-suh-leys).1 The adrenal glands need 21-hydroxylase to make proper amounts of hormones. This type of CAH is sometimes referred to as 21-hydroxylase deficiency. In CAH due to 21-hydroxylase deficiency, the adrenal glands cannot make enough cortisol or aldosterone. In addition, the glands make too much androgen. People with 21-hydroxylase deficiency also may not produce enough adrenaline.4
A small number of cases of CAH are caused by deficiency in a substance similar to 21-hydroxylase, called 11-hydroxylase.1 This type of CAH is sometimes referred to as 11-hydroxylase deficiency. In CAH due to 11-hydroxylase deficiency, the adrenal glands make too little cortisol and too many androgens. This type of CAH does not result in aldosterone deficiency.
Other very rare types of CAH include 3-betahydroxy-steroid dehydrogenase deficiency, lipoid CAH, and 17-hydroxylase deficiency. They are not discussed here.
CAH can be categorized as classic or nonclassic types based on severity:
- Classic CAH is more severe than the nonclassic form. It can be life threatening in newborns if it is not diagnosed. Classic CAH can be caused by either 21-hydroxylase or 11-hydroxylase deficiency.
- Nonclassic CAH is sometimes called late-onset CAH. It is a milder form of the disorder that usually is diagnosed in late childhood or early adolescence. Sometimes, people have nonclassic CAH and never know it. This form of CAH is almost always caused by 21-hydroxylase deficiency.
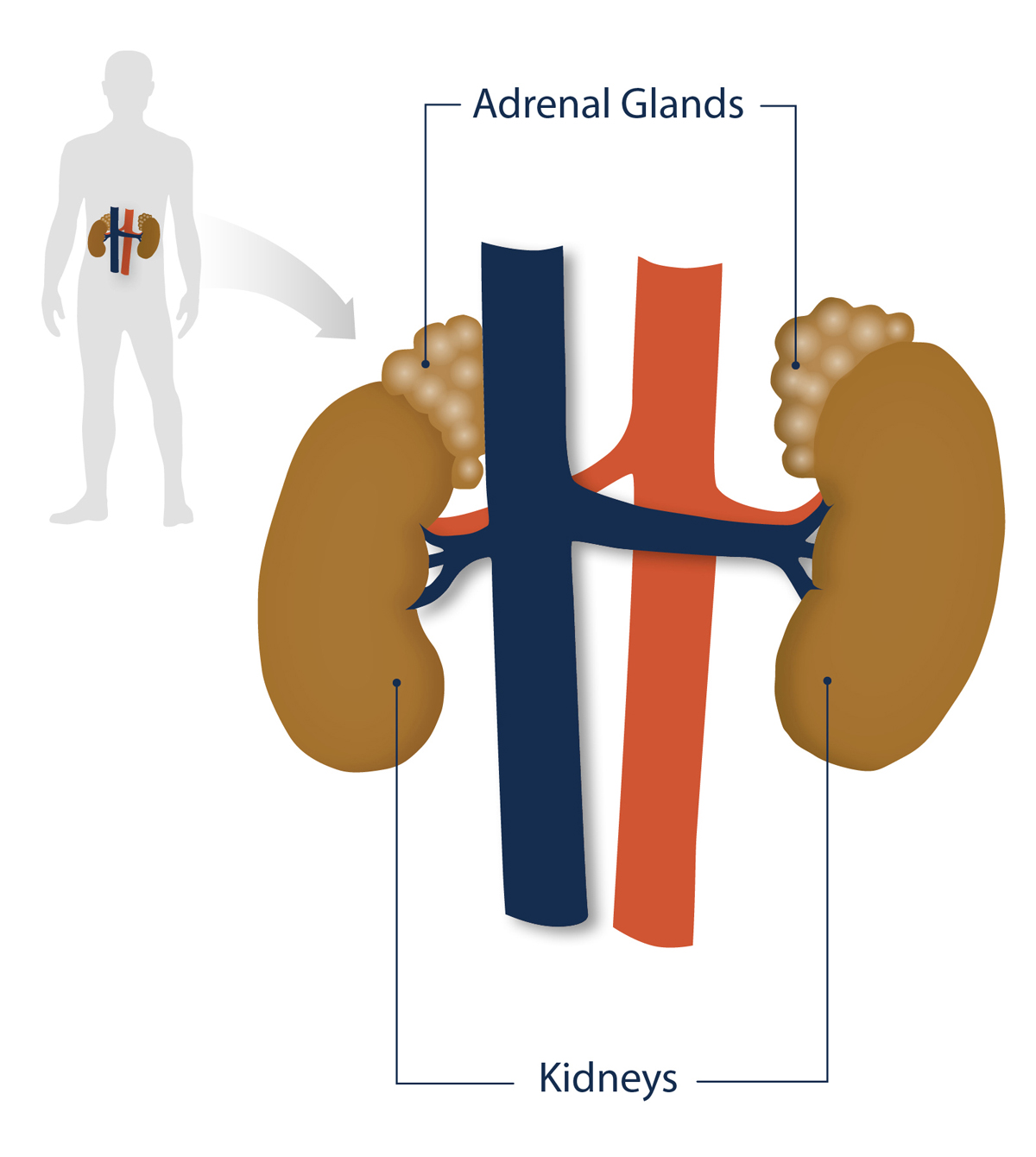
Figure 1. Position of the adrenal glands and kidneys in the human body.
Citations
- Speiser, P.W. and White, P.C. (2003). Congenital Adrenal Hyperplasia. New England Journal of Medicine, 349, 776-788. Retrieved on March 2, 2018, from http://www.nejm.org/doi/full/10.1056/NEJMra021561

- Genetic and Rare Diseases Information Center. (n.d.). Congenital Adrenal Hyperplasia. Retrieved May 2020 from https://rarediseases.info.nih.gov/diseases/1467/congenital-adrenal-hyperplasia
- Texas Department of State Health Services. (2001). Congenital adrenal hyperplasia: A handbook for parents. Retrieved June 26, 2012, from http://www.dshs.state.tx.us/newborn/hand_cah.shtm
- NICHD, NIH News Alert. (2006, February 19). People with common masculinizing disorder also lack adrenaline, NICHD study finds. Retrieved May 3, 2012, from http://www.nichd.nih.gov/news/releases/Pages/maledisorder.aspx
 BACK TO TOP
BACK TO TOP